利用 MISA-MD 进行级联碰撞模拟
基本原理
MISA-MD 作为一款分子动力学模拟软件,以粒子(分子/原子)作为研究对象,通过势函数计算粒子受力,并通过牛顿运动定律计算粒子的运动。
计算过程中通过时间步迭代的方式进行,在每一个时间步内,计算:
- 遍历每一个粒子,计算粒子所在位置的势能(通过截断半径范围内的所有粒子对该粒子所在位置的势能贡献计算);
- 依据势能,计算每一个粒子的受力(力等于势能的负梯度),这也是分子动力学的核心部分;
- 针对每一个粒子,依据其受力,采用牛顿第二定律获得其加速度; 再依据对应的时间步长,更新粒子的速度; 最后计算该时间步长内粒子的位移并更新粒子的位置。 但实际上,还需要考虑到如何减少数值误差,MISA-MD 中采用了精度更高的蛙跳法来进行牛顿运动方程的积分求解。
在并行化方面,MISA-MD 采用空间划分的思想,将模拟体系的原子划分到各个处理器核上,每个CPU核计算其中一部分原子的受力、运动。
相关概念
- 截断半径
分子动力学(Molecular Dynamics,MD)按受力范围可分为短程 MD 和长程 MD。在短程 MD 中,有截断半径的概念。
如果两个粒子距离大于截断半径,其之间的受力几乎为0,可以不考虑。
故此,短程 MD 中,计算粒子受力时,只需要考虑截断半径范围内的粒子对力的贡献即可。
对于金属材料体系,原子的受力基本都是短程力,采用的对应 MD 方法也是短程 MD。
MISA-MD 是一款应用于金属材料模拟的短程 MD 程序。 - 势函数
在分子动力学中,势函数是一个很重要的概念。势函数确定了粒子的受力,决定了模拟是否准确。势函数有很多种,例如简单的 L-J 势函数,复杂的金属体系中的 EAM 势函数。
对于金属材料,EAM 势函数是一种常用的势函数。由于其考虑到了电子的影响,能够很好地计算体系中的势能。
目前 MISA-MD 仅支持 Fe-Cu-Ni 的三元体系()后续会提供任意金属元素的支持,对应的势函数可以从 Interatomic Potentials Repository 获取到(Fe-Cu-Ni 体系的势函数见:https://www.ctcms.nist.gov/potentials/system/Cu-Fe-Ni/)。
注:之前示例中已经包括了该势函数文件:FeCuNi.eam.alloy。 - 级联碰撞
级联碰撞的概念是反应堆环境中用到的。对于裂变堆的压力容器材料(主要成分是Fe)或者聚变堆的钨材料,里面的原子会受到中子辐射,导致材料受到损伤。
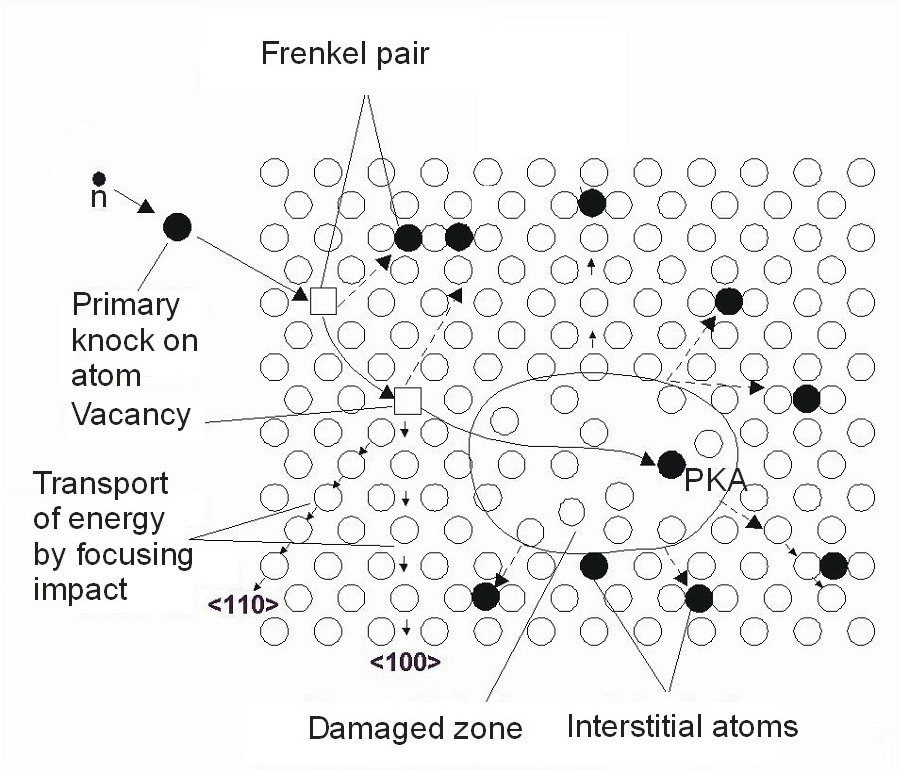
在宏观上的表现是材料的脆化、断裂等现象。在微观原子尺度上,初始材料中的原子是规整排列的,所有原子都在晶格上面(如下图);
但是反应堆环境下,可能某个原子受到中子撞击,将其撞离晶格点并将能量传递到这个原子上。一般我们将这个受到撞击的原子称为 PKA (Primary Knocked-on Atom)。
这个 PKA 原子还会接二连三地继续撞击周围其他原子,直到其能量都分散开来。这个过程就叫级联碰撞(如下图2)。
原子受到撞击,离开其原始的晶格点,原始的晶格点位置形成空位,离开晶格点的原子会形成间隙。这些空位和间隙在材料中都被称为缺陷,随着演化,影响到材料的宏观性能。
MISA-MD 可以对反应堆中的级联碰撞过程进行模拟,也是目前该程序的一个主要应用对象。
- 变时间步长 和 模拟阶段
这个概念是 MISA-MD 中采用的。一般地,在级联碰撞模拟的初期,PKA 原子速度会比较大,模拟的时间步长需要设置得比较小,这样牛顿运动方程的积分才会准确。 随着模拟的进行和体系的稳定,可以适当地增大时间步长,以加快模拟 (例如需要模拟到某个物理时间,可以增大时间步长以减少时间步数)。
所以,在 MISA-MD 中,设计了多个模拟阶段,不同模拟阶段可以给各个参数设置不同值,这些参数中就包括时间步数和时间步长。
MISA-MD 输入配置
具体的关于配置文件中每一个参数的详细解释和类型,可参考文档中的配置项说明。
MISA-MD 所有的输入配置都是在配置文件中进行指定的,以下对一些主要配置进行说明:
配置模拟体系
- 模拟 box 的大小:在配置文件中通过
simulation.phasespace选项指定模拟的三维空间的晶格数。 由于晶格是 BCC 结构的,所以模拟的原子数为 2 * x * y * z,其中 x,y,z 分别是simulation.phasespace选项指定的三个维度的晶格边长。例如设置simulation.phasespace选项为[80,80,80],则模拟的原子数为 1024000。 - 势函数文件: 通过
potential.file_path选项设定势函数文件路径。 - 合金比例: 通过
creation.alloy.ratio选项指定,详见配置项说明。例如设置的Fe,Cu,Ni 的比例为100:3:1,则MISA-MD程序即按照该比例初始原子类型。
配置模拟阶段和变时间步长
在级联碰撞模拟中,一般地,我们将模拟划分为四个阶段(MISA-MD 本身支持任意个数的阶)。
- 第一阶段为rescale (弛豫)过程。该阶段通过弛豫使得体系稳定。即每隔一定时间步重置体系的温度。下面的示例中,第一个阶段是每隔1个时间步将体系温度重置为 600K。该阶段的时间步数为500~1000左右,时间步长建议取0.001 ps。
- 第二阶段为碰撞阶段,生成 PKA。该阶段设置 PKA 能量、PKA 位置和 PKA 的速度分布,并在碰撞后继续往后模拟一段时间使得体系达到不剧烈变化状态。
- 后面一般我们还可以通过两个阶段的模拟,来使得体系稳定下来。这两个阶段的时间步长可以相比于前一个阶段长些。 可参照下面示例配置后两个阶段的时间步和步长,继续模拟大约30000步,体系基本能够达到稳定。
## MISA-MD 多阶段示例(配置文件的部分)
stages:
- name: rescale
step_length: 0.001
steps: 1000
rescale: # rescale to a temperature.
t: 600
every_steps: 1 # rescale every n steps
- name: collision
step_length: 0.0001
steps: 10000
set_v:
collision_step: 2 # unsigned long type, step relative to current stage, not global steps.
lat: [40, 40, 40, 0] # int array type
energy: 15000.0 # double, unit: eV, default: 0
direction: [1.0, 3.0, 5.0] # double array type, pka direction
- name: relax
step_length: 0.0005
steps: 10000
- name: run
step_length: 0.001
steps: 20000
配置级联碰撞
在模拟的碰撞阶段(第二个阶段),可以配置级联碰撞的参数。
该阶段的时间步长一般比较小,例如 0.0001 ps (如果能量高于30 kEv,步长还可以设置为 0.00005ps);时间步数一般大于10000步。
PKA 相关的参数设置说明如下:
- PKA 能量通过
set_v.energy指定,单位为电子伏特。实际裂变堆中,PKA 能量一般不超过 50 KeV。 - PKA 位置(通过
set_v.lat指定)一般可以设置为位于模拟box中间(例如模拟box为[80,80,80],则PKA位置可以设置为[40, 40, 40, 0](第四个参数0无实际意义) )。 - PKA 速度方向通过
set_v.direction设置,例如速度方向设置为[1.0, 3.0, 5.0],则 PKA 速度和向量⟨1, 3, 5⟩平行。
模拟结果后处理
采用 md-tools 工具进行后处理,将MISA-MD输出的二进制文件转化为原子坐标格式(.xyz 格式)。详情参见 md-tools 小节。
模拟结果分析与可视化
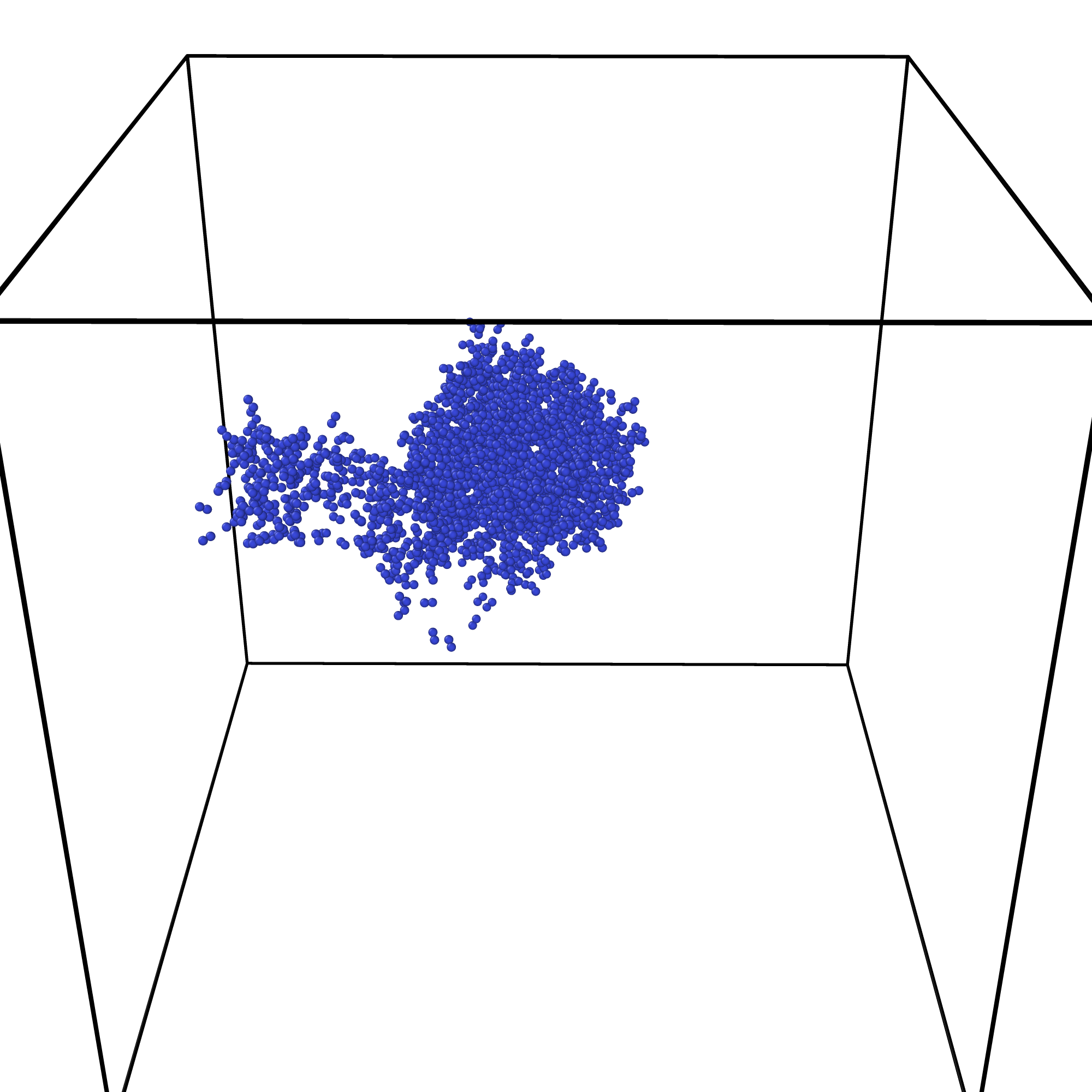
我们可以采用 ovito 工具进行结果分析,主要分析 MD 模拟过程中的缺陷演化过程。 采用 ovito 工具自带的 Wigner-Seitz 缺陷分析方法,可以得到每个输出时间步的缺陷数量和缺陷的位置信息。
如下图即是其中一个输出时间步的可视化结果。